
Vertuschter Skandal
Text- Größe: 430 S.
- Kategorie: Angewandte Bereiche der MedizinBearbeiten
Um den Sachverhalt zu klären, hatte das Ministerium für Gesundheitswesen eine Expertenkommission unter dem Vorsitz von Friedrich Oberdoerster eingesetzt. Diese tagte am 18. Januar 1979 im Staatlichen Kontrollinstitut für Seren und Impfstoffe Berlin. Das Staatliche Kontrollinstitut repräsentierten neben dem Leiter vier weitere Mitarbeiter. Anwesend waren Vertreter der Bezirksinstitute für Blutspende- und Transfusionswesen Cottbus, Halle (Saale) und Magdeburg. Auch Vertreter mehrerer Kliniken nahmen an der Sitzung der Expertenkommission teil. So war ein Vertreter des Städtischen Krankenhauses Berlin Prenzlauer Berg, der Frauenklinik Dresden und der Medizinischen Klinik der Medizinischen Akademie Dresden anwesend. Auch eine Mitarbeiterin der Hauptabteilung Hygiene und Staatliche Hygieneinspektion des Gesundheitsministeriums war anwesend, ebenso wie je eine Mitarbeiterin des Hygieneinstituts Erfurt und des Instituts für Arzneimittelwesen der DDR.108
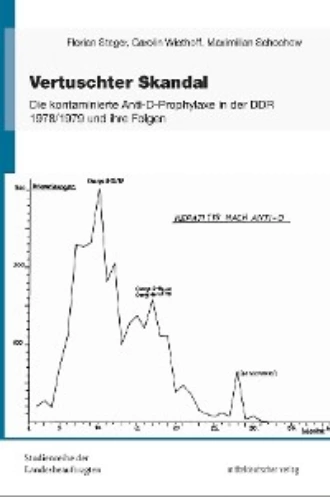
Bis zum 19. Januar 1979 waren 47 Erkrankungen bekannt geworden. Diese führte die Expertenkommission auf die Verwendung der beiden Spenderplasmen aus Neubrandenburg in den Chargen 8 bis 15 zurück. Aufgrund der durchgeführten Untersuchungen wurde davon ausgegangen, dass es sich nicht um eine Hepatitis B, sondern um eine Hepatitis A oder Non-A-Non-B handelte. Zum Nachweis der Hepatitisform sollten mehrere Maßnahmen ergriffen werden. Ein elektronenmikroskopischer Nachweis von Antikörpern gegen Hepatitis A sollte in den Seren und Stuhlproben von 10 frisch erkrankten Patienten erbracht werden. Zur Sicherung einer Non-A-Non-B-Hepatitis wurde vorgeschlagen, ein Referenzlaboratorium der Weltgesundheitsorganisation einzubeziehen. Es war vorgesehen, die Spenderseren und Blutproben einiger erkrankter Patientinnen in mehreren Testverfahren zu untersuchen, um eine Hepatitis B wirklich auszuschließen.109
Bei der Verwendung der Chargen 17 bis 23 ging die Expertenkommission von einem „vertretbarem Risiko hinsichtlich der Gefahr der Übertragung einer Hepatitis“ aus. Sie begründete dies mit der Tatsache, dass diese Chargen kein Plasma der beiden Spender aus Neubrandenburg enthielten. Zudem sei „anzunehmen, daß durch die Sterilisation der wiederholt verwendeten Austauschgele ggf. vorhandenes Hepatitisvirus inaktiviert“ werde. Eine in Betracht gezogene Strahlensterilisation der Chargen 8 bis 15 lehnte die Expertenkommission ab. Gleichzeitig wurde „dringend empfohlen“, die Immunprophylaxe nicht zu unterbrechen.110 Alle bisher nicht erkrankten Frauen, die mit dem Anti-D dieser Chargen behandelt worden waren, sollten im Abstand von vier Wochen zweimal je 6 ml Gammaglobulin erhalten und in die Dispensairebetreuung einbezogen werden. Auch Säuglinge, deren Mütter an Hepatitis erkrankt waren oder hohe Transaminasewerte aufwiesen, sollten Gammaglobulin erhalten. Die Erfassung und Betreuung der Patientinnen sollte den Kreisärzten übertragen werden.111
Neben dem Einsatz einer Expertenkommission zur Klärung der Ursachen hatte das Ministerium für Gesundheitswesen weitere Maßnahmen ergriffen. Gesundheitsminister Ludwig Mecklinger informierte die Bezirksärzte am 23. Januar 1979 darüber, dass die Chargen 8 bis 15 gesperrt und sichergestellt worden waren. Er gab an, dass der Zusammenhang zwischen den aufgetretenen Erkrankungen und der Anti-D-Prophylaxe untersucht werde und wies spezielle Maßnahmen zum Umgang mit den betroffenen Frauen an.112
Neben dem Bezirksinstitut in Halle (Saale) wurde auch das Bezirksinstitut in Neubrandenburg geprüft, in dem am 9. Februar 1979 ein „Informationsbesuch“ von Vertretern des Kontrollinstituts stattfand. Dabei stellte sich heraus, dass die Erythrozyten der Antigen-Spenderin nur einmalig bei den fünf Spendern verwendet worden waren. Zwar war die Spenderin seit 1976 im Bezirksinstitut Neubrandenburg registriert und hatte vor diesem Zeitpunkt am 15. November 1977 das letzte Mal Blut gespendet. Ihre Erythrozyten waren jedoch nur deshalb benutzt worden, weil die bisherige Spenderin erkrankt war, die seit 1970 Erythrozyten gespendet hatte. Die Spenderin, mit deren Erythrozyten die fünf Plasmaspender immunisiert worden waren, hatte sich bereits Mitte Januar 1978 aufgrund von Oberbauchbeschwerden in ärztliche Behandlung begeben. Stuhl und Urin seien unauffällig gewesen.113 Die Erythrozytenspenderin war am 14. April 1978 in das Bezirksinstitut für Blutspende- und Transfusionswesen Neubrandenburg bestellt und anschließend zur weiteren Klärung der Diagnose in das Bezirkskrankenhaus überwiesen worden. Eine erbetene Leberpunktion hatte nicht stattgefunden, da im Bezirkskrankenhaus Neubrandenburg „weder klinisch noch paraklinisch ein Anhalt für eine Hepatitis zu finden war.“114
Diese Information hatte der Ärztliche Direktor des Neubrandenburger Bezirksinstituts bereits Ende Januar 1979 Oberdoerster mitgeteilt und ihm die Epikrisen und einen kurzen Bericht zugesandt. Alle fünf Personen hätten mehrfach Blut gespendet und seien niemals bei den durchgeführten Kontrolluntersuchungen aufgefallen. Er informierte Oberdoerster zudem über die negativen Testergebnisse hinsichtlich des HBs-Antigens sowie des HBs-Antikörpers, die nach der Prüfung in Dessau, Erfurt und Magdeburg vorlagen.115 Die Virushepatitis aller fünf Spender war durch eine Biopsie gesichert. Bei einem Spender hatte die Hepatitis einen anikterischen Verlauf genommen. Im Protokoll wurde zudem über das gewonnene Material und die Lieferung berichtet. Das Bezirksinstitut in Halle (Saale) hatte mehr Blut von der Spenderin als von dem Spender erhalten. Das Blut der Erythrozytenspenderin war „bis auf die zur Immunisierung verwendeten Blutkörperchen“ vernichtet worden.116 Ferner ging es um die zukünftige Produktion des Anti-D-Immunglobulins im Bezirksinstitut für Blutspende- und Transfusionswesen Neubrandenburg. Das Material sollte trotzdem den Vermerk „staatlich geprüft im BIBT-Halle“ erhalten.117 Das Institut für Arzneimittel sollte darüber entscheiden, ob die staatliche Prüfung in Zukunft durch das Bezirksinstitut in Halle (Saale) übernommen werden könne. Im Rahmen des Informationsbesuchs war offenbar auch der Verdacht geäußert worden, dass die Spender vor ihrer Spende nicht den Vorschriften entsprechend untersucht worden waren.118 Zwar hatte der Ärztliche Direktor des Neubrandenburger Instituts zuvor auf mehrere Tests hingewiesen, doch diese bezogen sich auf den Zeitraum nach der Spende und der Erkrankung.119 Er teilte dem Kontrollinstitut im Februar 1979 mit, dass seine „diesbezüglichen Nachforschungen“ ergeben hätten, dass die Vermutung „leider den Tatsachen entspricht.“ Lediglich das Blut der Erythrozytenspenderin war am 27. Januar 1978 einem Hepatitis-Screening zugeführt worden. Der Ärztliche Direktor musste zugeben, dass dieses Vorgehen „nicht den bestehenden gesetzlichen Bestimmungen“ entsprach und drückte sein Bedauern darüber aus. Bis dahin sei er stets davon überzeugt gewesen, dass die notwendigen Kontrolluntersuchungen durchgeführt würden. Er versicherte, dass dies auch ab sofort der Fall sei.120
Über diese ersten Ergebnisse informierte Oberdoerster das Ministerium für Gesundheitswesen, das ihn zuvor offenbar um eine Einschätzung gebeten hatte. Oberdoerster fasste den Verlauf der Ereignisse knapp zusammen und teilte die Ergebnisse der Betriebskontrolle im Bezirksinstitut für Blutspende- und Transfusionswesen Halle (Saale) mit. Er sah insbesondere die Gütevorschrift Anti-D-Immunglobulin verletzt, nach der Plasmaspender den Forderungen zur Anordnung über den Blutspende- und Transfusionsdienst beziehungsweise den hierzu erlassenen Richtlinien entsprechen mussten. In Richtlinie Nr. 1 war festgelegt, dass die Spender frei von übertragbaren Krankheiten sein mussten. Nicht in Frage kamen dabei Personen, die in den letzten fünf Jahren vor der Spende an einer Hepatitis erkrankt waren. Auch wenn in deren unmittelbaren Umgebung im letzten halben Jahr vor der Blutspende eine Hepatitis aufgetreten war, sollten potenzielle Spender nicht herangezogen werden. Die Richtlinie sah ferner die Erstuntersuchung des Spenders vor. Bei dieser sollten eine Anamnese, ein Siebtest auf Hepatitis und die Kontrolle des Sozialversicherungsausweises stattfinden, vor jeder weiteren Blutspende eine Nachuntersuchung.121 Dass das Bezirksinstitut für Blutspende- und Transfusionswesen Neubrandenburg diese Richtlinien nicht beachtet hatte, erwähnte Oberdoerster nicht. Stattdessen ging er ausführlich auf Schuberts Verantwortlichkeit ein, der seiner Ansicht nach selbst ein Risiko in der Verwendung der Chargen gesehen habe. Oberdoerster sah in den Festlegungen der Gütevorschrift und der Richtlinie Nr. 1 ausreichende Gründe gegen den Einsatz der beiden Chargen. Diese Vorschriften hätten auch bei der weiteren Verwendung der Plasmen herangezogen werden müssen.
Oberdoerster kritisierte, dass Schubert davon ausgegangen war, mithilfe des Fraktionierungsverfahrens Hepatitisviren zu eliminieren, und sich nur auf Testverfahren zum Nachweis des HBs-Antigens gestützt hatte. Er wandte dagegen ein, dass 70 bis 90 % aller posttransfusioniellen Hepatitiden durch Non-A-Non-B-Hepatitisviren ausgelöst werden und stützte sich dabei auf einen Bericht der Weltgesundheitsorganisation. Die Hepatitissicherheit der Immunglobuline werde zwar in der Literatur häufig erwähnt und auch von Expertengruppen der Weltgesundheitsorganisation betont. Doch um diese zu gewährleisten, seien mehrere Faktoren wichtig. Hierzu gehörten unter anderem die Kriterien für die Auswahl der Plasmaspender, das Fraktionierungsverfahren und die Anzahl der Plasmen als Ausgangsmaterial für eine Charge. Oberdoerster bezifferte diese bei normalem Human-Immunglobulin auf Plasmen von 1.000 und mehr Spender.122 Er kritisierte insbesondere, dass sich Schubert bei der Fraktionierung nur auf die Wirkung des Äthanols verlassen habe, wofür er keinen Anhaltspunkt sah. Stattdessen verwies er darauf, dass allgemein eine Wärmebehandlung von Albuminlösungen zur Inaktivierung gegebenenfalls vorhandener Hepatitisviren gefordert werde. Schubert hatte seiner Meinung nach keine Kenntnisse über diese Kriterien oder hatte diese fahrlässig missachtet. Der Vollständigkeit halber fügte Oberdoerster hinzu, dass Schubert seine Entscheidung auch in Anbetracht der Schwierigkeit, ausreichend Material zu erhalten, getroffen hatte.
Bezüglich der Verantwortlichkeit des Leiters der Technischen Kontrollorganisation schätzte er ein, dass dieser den Einsatz der Plasmen für die Chargen 8 bis 14 hätte verhindern können, wenn er die Unterlagen dazu und deren Verbleib geprüft hätte. Eine Überprüfung wäre gerade bei diesen Plasmen wichtig gewesen, da es keine Prüfmethode zum Ausschluss einer Kontamination durch Hepatitisviren gebe. Oberdoerster hielt dem Leiter der Technischen Kontrollorganisation vor, dass dieser beim Antrag auf Freigabe der Charge 15 keinen entsprechenden Hinweis gegeben hatte. Nach Punkt 26 der Gütevorschrift hätte ihm dieser Abweichungen von der Herstellungsvorschrift sofort mitteilen müssen.123
Aufgrund der „Versäumnisse der beiden Verantwortlichen“ sei es zur Erkrankung von mehr als 250 jungen Frauen gekommen.124 Oberdoerster wollte diese Stellungnahme auf der nächsten Sitzung der Expertenkommission vorlegen, die am 28. Februar 1979 im Berliner Institut für Angewandte Virologie stattfand. Vertreter des Bezirksinstituts für Blutspende- und Transfusionswesen Halle (Saale) waren nicht mehr zugegen. Ansonsten waren mit einer Ausnahme dieselben Teilnehmer wie auf der ersten Sitzung im Januar anwesend.125 Im Protokoll war festgehalten, dass die Anwesenden Oberdoersters Einschätzung teilten.
Die auf der Sitzung anwesende Vertreterin des Ministeriums für Gesundheitswesen berichtete, dass etwa 4.000 Ampullen der gesperrten Chargen Anti-D zur Anwendung gekommen waren. Die Differenz zwischen den Summen der Chargengrößen und der Rückläufe der gesperrten Chargen wurde mit 4.363 Ampullen beziffert. Bis zum 21. Februar waren etwa 2.100 Enzymuntersuchungen bei den betroffenen Frauen durchgeführt worden, von denen etwa die Hälfe erhöhte Werte gezeigt hatte. Das Ministerium für Gesundheitswesen rechnete mit einer Erkrankung von 1.000 bis 1.500 weiteren Personen. Mecklinger hatte angewiesen, die Enzymuntersuchungen auf alle Säuglinge auszudehnen, deren Mütter mit den gesperrten Chargen immunisiert worden waren. Gründe hierfür waren die Erkrankung von zwei Säuglingen im Bezirk Leipzig und erhöhte Enzymwerte bei vier Säuglingen in den Bezirken Berlin, Potsdam und Suhl.126
Bislang waren keine Untersuchungen zum Nachweis von Antikörpern gegen Hepatitis A durchgeführt worden. Dies teilte der Vertreter der Medizinischen Klinik der Medizinischen Akademie Dresden mit. Mit deren Auftreten sei erst sechs bis acht Wochen nach dem Ausheilen der Krankheit zu rechnen. Er gab an, dass als „infektiöses Agens“ weiterhin das Non-A-Non-B-Hepatitisvirus für wahrscheinlich gehalten werde.127 In zehn Fällen hatte die histologische Untersuchung das typische Bild einer Virushepatitis ergeben. Weitere Leberbiopsien sollten aber nicht mehr vorgenommen werden. Stattdessen thematisierte der Vertreter der Medizinischen Klinik der Medizinischen Akademie Dresden die Bedeutung der Nachkontrolle aufgrund der Möglichkeit einer chronischen Erkrankung. Er schlug vor, ein einheitliches Schema für die Erfassung der Befunde und die Nachkontrolle anzuwenden. Dieses sollte zwischen ihm, dem Ministerium für Gesundheitswesen und der Chefärztin der Klinik Berlin Prenzlauer Berg abgestimmt werden.128
Diese unterbreitete gemeinsam mit dem Vertreter der Medizinischen Klinik der Medizinischen Akademie Dresden Vorschläge in Bezug auf die Nachsorge. Bei Patientinnen ohne Symptome (1. Gruppe) sollte nach zwei negativen Enzymbefunden ein dritter Siebtest in der 15. Woche durchgeführt werden. Bei einem unkomplizierten Krankheitsverlauf (2. Gruppe) sollte vier Wochen nach der Behandlung ein Enzymtest durchgeführt werden. Bei negativem Ergebnis war der Test nach weiteren sechs Monaten zu wiederholen. Falls der Test positiv war, sollte die Patientin in die dritte Gruppe (komplizierter Krankheitsverlauf) eingestuft werden. Diese Patientinnen sollten in die Dispensairebetreuung aufgenommen und Enzymuntersuchungen in dreimonatigen Abständen bis zur Ausheilung unterzogen werden.129 Diese Vorschläge waren die Grundlage für eine erneute Weisung des Ministers für Gesundheitswesen am 2. März 1979, welche die Nachuntersuchungen regelte. Mecklinger orientierte sich darin genau an den Vorschlägen der Expertenkommission.130
Die Vertreterin des Hygieneinstituts Erfurt berichtete auf der Sitzung der Expertenkommission, dass bei einer Untersuchung von 72 Frauen in drei Fällen das HBs-Antigen nachgewiesen werden konnte. Sie schloss aber nicht aus, dass diese Patientinnen bereits vor der Immunprophylaxe Trägerinnen des Antigens gewesen waren. Gleichzeitig wies sie darauf hin, dass „im Bezirk Erfurt in vereinzelten Fällen auch nach der Sperrung HIG [Human-Immunglobulin]-Anti-D der gesperrten Chargen zur Anwendung gekommen“ sei.131 Der Leiter des Bezirksinstituts für Blutspende- und Transfusionswesen Magdeburg ging explizit auf die Verantwortlichkeit von Schubert und dem Leiter der Technischen Kontrollorganisation ein. Er schloss sich Oberdoersters Einschätzung an. Auch ein Engpass an Ausgangsmaterialien sei kein Grund, ein derartiges Risiko einzugehen. Die bisherige Konzeption der Rhesus-Prophylaxe in der DDR kritisierte er als „falsch“ und schlug vor, „ein i. m. applizierbares“ Anti-D-Immunglobulin herzustellen.132 Damit könnten auch Ausgangsplasmen mit einem niedrigeren Gehalt an Antikörpern gegen die Blutgruppeneigenschaft D zur Verarbeitung gelangen. Mehrere Personen, die künstlich immunisiert worden waren, kämen dann als Plasmaspender infrage. Bisher betraf dies nur zwei bis drei Personen von 30. Dementsprechend könne das Ausgangsmaterial pro Charge aus einem größeren Pool von Plasmen zusammengestellt und die Produktion des Anti-D-Immunglobulins in das Institut für Impfstoffe Dessau überführt werden, „wo eine entsprechende Technologie bereits vorhanden“ sei. Die Vertreterin des Bezirksinstituts für Blutspende- und Transfusionswesen Cottbus unterstützte diesen Vorschlag: „Die bisherige Weigerung des Instituts für Impfstoffe Dessau wegen fehlender Fraktionierungskapazität vor 1983 die Produktion zu übernehmen“, könne nicht akzeptiert werden.133
Zu den auf der Beratung besprochenen Themen wurden Schlussfolgerungen festgelegt. Ein erster Punkt betraf den Fachausschuss für Blutspende- und Transfusionswesen. Dieser sollte vom Minister für Gesundheitswesen mit einer Kontrolle der Einrichtungen des Blutspende- und Transfusionswesen beauftragt werden. Hierzu gehörten insbesondere die Auswahl und gesundheitliche Überwachung der Antigen- und Plasmaspender und die anhand der Plasmen durchgeführten labordiagnostischen Untersuchungen. Auch die Aufgaben der Immunisierungskommission und die Erarbeitung neuer Richtlinien sollten diesem Fachausschuss übertragen werden. Diese betrafen die Auswahl der Antigenspender, der Plasmaspender, die Aufbewahrung des Antigens bis zur Verwendung und die Vorratsbildung des Wirkstoffs. Ziel war es, die Plasmen sechs Monate vor der Verwendung aufzubewahren und damit die Inkubationszeit der Hepatitis abzuwarten.134 Durch den Fachausschuss für Blutspende- und Transfusionswesen sollte ferner eine Studie erarbeitet werden, ob mit dem in Dessau erprobten Fraktionierungsverfahren auch Anti-D-Immunglobulin zur intramuskulären Anwendung hergestellt werden könne.
In die überarbeitete Hepatitisrichtlinie sollten weitere Verpflichtungen des behandelnden Arztes aufgenommen werden. Hierzu gehörte, dass er eine ihm bekannt gewordene Hepatitiserkrankung eines Spenders von Blut oder Blutbestandteilen unverzüglich dem zuständigen Bezirksinstitut für Blutspende- und Transfusionswesen zu melden hatte. Gleichzeitig sollte das Institut für Impfstoffe Dessau nicht nur das Immunplasma vor der Mischung zum Ausgangspool auf HBsAg testen, sondern auch die von der Weltgesundheitsorganisation empfohlene zehnstündige Wärmebehandlung der Albuminlösungen bei 60 Grad Celsius durchführen.135
Im März 1979 wurde bekannt, dass auch nach der Verwendung nachfolgender Chargen Erkrankungen aufgetreten waren. Es handelte sich hierbei um die Chargen 16 und 17. Daraufhin überprüften Mitarbeiter des Staatlichen Kontrollinstituts für Seren und Impfstoffe das Institut in Halle (Saale) am 12. März 1979 erneut. Dieses Mal war auch Oberdoerster persönlich anwesend. Bei der Kontrolle wurde festgestellt, dass in den Produktionskontrollen zur Herstellung der Chargen 16, 17, 18 und 23 die Nutzung der Waschflüssigkeit der vorherigen Charge nicht ausgewiesen war. Für die Chargen 19, 20, 21 und 22 war dies hingegen ersichtlich, ebenso für die 1979 produzierten Chargen 2 bis 7. Die Waschflüssigkeit der Charge 23 war hingegen nicht in die erste Charge des Jahres 1979 gelangt, da diese zu diagnostischen Zwecken an die Laborabteilung übergeben worden war.136 Die Angaben in der Produktionsdokumentation zu den Chargen 16 bis 23 wurden als unvollständig betrachtet. Es müsse davon ausgegangen werden, dass „wie üblich“ auch diese Chargen die Waschflüssigkeit der jeweils vorangegangenen Charge enthielten.137 Damit sei die während der Betriebskontrolle im Januar getroffene Feststellung, dass bei Charge 16 eine Ausnahme vorlag, unzutreffend.
Oberdoerster ließ daraufhin die Chargen 16 bis 23 sperren. Noch vorhandene Zwischenprodukte der Charge 23 sollten sofort aus dem Fraktionierungsbereich entfernt werden. Ein erhöhtes Hepatitisrisiko für die Chargen aus 1979 wurde aber ausgeschlossen. Das Kontrollinstitut erteilte dem Bezirksinstitut in Halle (Saale) mehrere Auflagen. Die zurückgeführten Anteile der gesperrten Chargen 16 bis 22 waren in ihrer Originalverpackung unter Verschluss zu nehmen. Die Charge 23 war noch nicht ausgeliefert worden. Die Waschflüssigkeit und vorhandene Zwischenprodukte dieser Charge sollten vernichtet und das Vernichtungsprotokoll an das Kontrollinstitut gesendet werden. Ab Charge 7 aus dem Jahr 1979 durfte die anfallende Waschflüssigkeit nicht mehr in nachfolgende Chargen eingehen. Der Verbleib der Flüssigkeiten musste im Produktionsprotokoll ausgewiesen werden. Außerdem war die Produktionsdokumentation zukünftig so zu gestalten, „daß die vorhandenen Angaben Fehlinterpretationen nicht zulassen.“138 Eine neue Vereinbarung zwischen dem Direktor des Produktionsbetriebs in Halle (Saale) und den zuliefernden Einrichtungen des Blutspende- und Transfusionswesens war vorgesehen. Jedes gelieferte Einzelplasma sollte künftig ein Zertifikat enthalten. Anhand dessen sollte der Leiter der Technischen Kontrollorganisation des Bezirksinstituts Halle (Saale) erkennen, dass die Spender den Anforderungen gemäß den Richtlinien entsprachen.139
Zudem ergab die Kontrolle, dass 700 ml der Charge 15 verschwunden waren. Obwohl alle Lagermöglichkeiten kontrolliert wurden, blieb diese Teilcharge unauffindbar. Eine Mitarbeiterin des Instituts mutmaßte, dass sie möglicherweise zum Zeitpunkt der Abfüllung der Charge 15 versehentlich bei der Reinigung anderer Glasgefäße verworfen worden war. Berechnungen wurden angestellt, die eine Verwendung der 700 ml unwahrscheinlich machten. Dennoch wollten die Kontrolleure nicht ausschließen, dass diese 700 ml als Teilcharge in einen anderen Produktionsschritt der nachfolgenden Chargen eingegangen waren.140
Einen Tag nach der Kontrolle berichtete Gesundheitsminister Mecklinger den Bezirksärzten über das Auftreten weiterer Erkrankungen nach der Verwendung der nachfolgenden Chargen. Und das, obwohl eine von ihm berufene Expertengruppe und das Staatliche Kontrollinstitut für Seren und Impfstoffe (SKISI) eine Kontamination dieser Chargen „nach gewissenhafter Einschätzung (…) weitgehend ausgeschlossen“ hatten.141 Im Zusammenhang mit den Chargen 16 und 17 handelte es sich bislang um jeweils drei Erkrankungen. Mecklinger hatte daher am 12. März 1979 die Sperrung der Chargen 16 bis 23 veranlasst. Die Anti-D-Prophylaxe sollte mit Chargen aus 1979 fortgeführt werden.142